CHAPTER 1
General Overview
This version of Molconn-Z was built from an all-new
library of Molconn-Z functionality that we have been developing over the
last 12-15 months. The previous core Molconn-Z program, which had become
difficult to work on and modify because of its structure, was written in
FORTRAN. The new core algorithms are written in standard C in a toolkit
structure. This enables us to much more quickly develop new Molconn-Z applications
to respond to user needs, and also allows us to distribute to interested
customers the means to develop their own Molconn-Z applications.
The Molconn-Z software is designed to carry out
the computation of a wide range of topological indices of molecular structure.
These indices represent important elements of the molecular structure information
which is useful in relating structure to properties. These variables of
molecular structure include (but are not limited to) the molecular connectivity
chi indices, m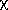 t and
mtv; kappa
shape indices, m
t and
mtv; kappa
shape indices, m and m
and m ;
electrotopological state indices, Si; hydrogen electrotopological
state indices, HESi; atom type and bond type electrotopological
state indices; new group type and bond type electrotopological state indices;
topological equivalence indices and total topological index; several information
indices, including the Shannon and the Bonchev-Trinajstiç information
indices; counts of graph paths, atoms, atoms types, bond types; and others.
;
electrotopological state indices, Si; hydrogen electrotopological
state indices, HESi; atom type and bond type electrotopological
state indices; new group type and bond type electrotopological state indices;
topological equivalence indices and total topological index; several information
indices, including the Shannon and the Bonchev-Trinajstiç information
indices; counts of graph paths, atoms, atoms types, bond types; and others.
These indices have been widely used in QSAR analyses
and other types of relationships between the structure of molecules and
their properties. Discussion of the definitions and background of the chi,
kappa, electrotopological state, and topological equivalence indices are
given in Chapter
2 along with appropriate references. Further, references are given
for several reviews of the development and use of topological indices at
the end of this chapter as well as in Chapter
2.
Installation and related instructions are
detailed in Chapter 3.
Molconn-Z is set up to be user-friendly and flexible.
See Chapter
4 for detailed information. Input of molecular structure is done with
one of 3 molecular structure file formats: Daylight
SMILES, MDL
(sdf), or Tripos
(mol2). These inputs are now handled using linked libraries from either
Daylight for SMILES (requires a license from Daylight) or OpenEye
Scientific Software (OElib) for SMILES,
sdf and mol2 file. See Chapter
5 for detailed information on the use of each of these input formats.
In addition to the most common forms of input,
Molconn-Z permits flexibility in output by changing a few keywords in the
control file, see Chapter
4 for detailed information. The output from Molconn-Z is placed in
a single output file with options for user control over the contents.
This file is compatible for use with the commonly
used statistical package SAS from The SAS Institute in Cary, NC., and should
be easily adapted for use with other statistical packages.
The general flow
of information for Molconn-Z is fairly simple - the command only accepts
3 arguments (first for the control file name, second for the input molecular
structure file name and third for output file name); it reads the control
file to determine run parameters and structure file type; it then reads
one molecule at a time and computes the descriptors. In general, the user
creates molecule structure either by use of a commercial package or, perhaps,
simply in an edited text (ASCII) file. The user also creates an ASCII
control file which defines the input molecular structure format and the
output format desired. Besides these two input files, the only other item
the user needs to worry about are the license files which are defined by
the environment variables DY_LICENSEDATA for the (optional) Daylight license
and MCONN_LICENSE for the (required) eduSoft/Molconn-Z license.
The user starts execution of Molconn-Z simply
by using the command line syntax:
Error messages, if any, are printed to the console
screen.
It should be noted that no special settings are
necessary with this new Molconn-Z to obtain optimal performance with large
databases.
Molconn-Z is designed to run on any UNIX/LINUX
platforms, as well as on microcomputers running Windows 95, 98, ME, 2000
or XP.
General References
1. L. B. Kier and L. H. Hall, Molecular Connectivity
in Structure-Activity Analysis, Research Studies Press, John Wiley
and Sons, Letchworth, England, (1986).
2. L. B. Kier and L. H. Hall, Molecular Connectivity
in Chemistry and Drug Research, Academic Press, New York, 1976.
3. L. H. Hall, "Computational Aspects of Molecular
Connectivity and its Role in Structure-Property Modeling" in Computational
Chemical Graph Theory, Chap. 8, pp 203-233, D. H. Rouvray, ed., Nova
Press, New York (1990).
4. L. B. Kier, "Indexes of Molecular Shape from
Chemical Graphs" in Computational Chemical Graph Theory, Volume
II, Chap. 6, pp 152-174, D. H. Rouvray, ed., Nova Press, New York (1990).
5. L. H. Hall and L. B. Kier, "The Molecular
Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property
Relations", in Reviews of Computational Chemistry, Chap. 9, pp 367-422,
Donald Boyd and Ken Lipkowitz, eds., VCH Publishers, Inc. (1991).
6. L. B. Kier and L. H. Hall, "An Atom-Centered
Index for Drug QSAR Models", in Advances in Drug Design, Vol. 22,
B. Testa, ed., Academic Press(1992).
7. A. T. Balaban, ed. Chemical Applications
of Graph Theory, Academic Press, New York, (1976).
8. N. Trinajstic', Chemical Graph Theory,
Vols. I, II, CRC Press, Boca Raton, FL, (1983).
9. R. B. King, ed., Chemical Applications
of Topology and Graph Theory, Amsterdam, (1983).
10. D. H. Rouvray, Am. Sci., 61,
729, (1973). The Search for Useful Topological Indices in Chemistry.
11. D. H. Rouvray, Sci. Am., 255,
40, (1986). Predicting Chemistry from Topology.
12. P. J. Hansen and P. C. Jurs, J. Chem.
Ed., 65, 574, (1988). Chemical Applications of Graph Theory
II: Isomer Enumeration.
13. A. Sabljic' and N. Trinajstic', Acta Pharm.
Jugosl., 31, 189, (1981). Quantitative Structure-Activity Relationships:
The Role of Topological Indices.